






医疗器械制造展Medtec谈医疗器械GMP对“空调净化系统”的要求
2024-08-16
01 前言
山东省医疗器械洁净室(区)现场检查指南(2021版)
02 GMP的要求
《医疗器械生产质量管理规范附录无菌医疗器械》、
《医疗器械生产质量管理规范附录体外诊断试剂》、
《医疗器械生产质量管理规范体外诊断试剂现场检查指导原则》、
《医疗器械生产质量管理规范无菌医疗器械现场检查指导原则》、
以上医疗器械GMP对空调系统的检查重点和检查方法进行了规定,在无菌医疗器械GMP检查指导原则中有一个2.2.1和2.9.1星号(*)项,其余均为一般不符合项。
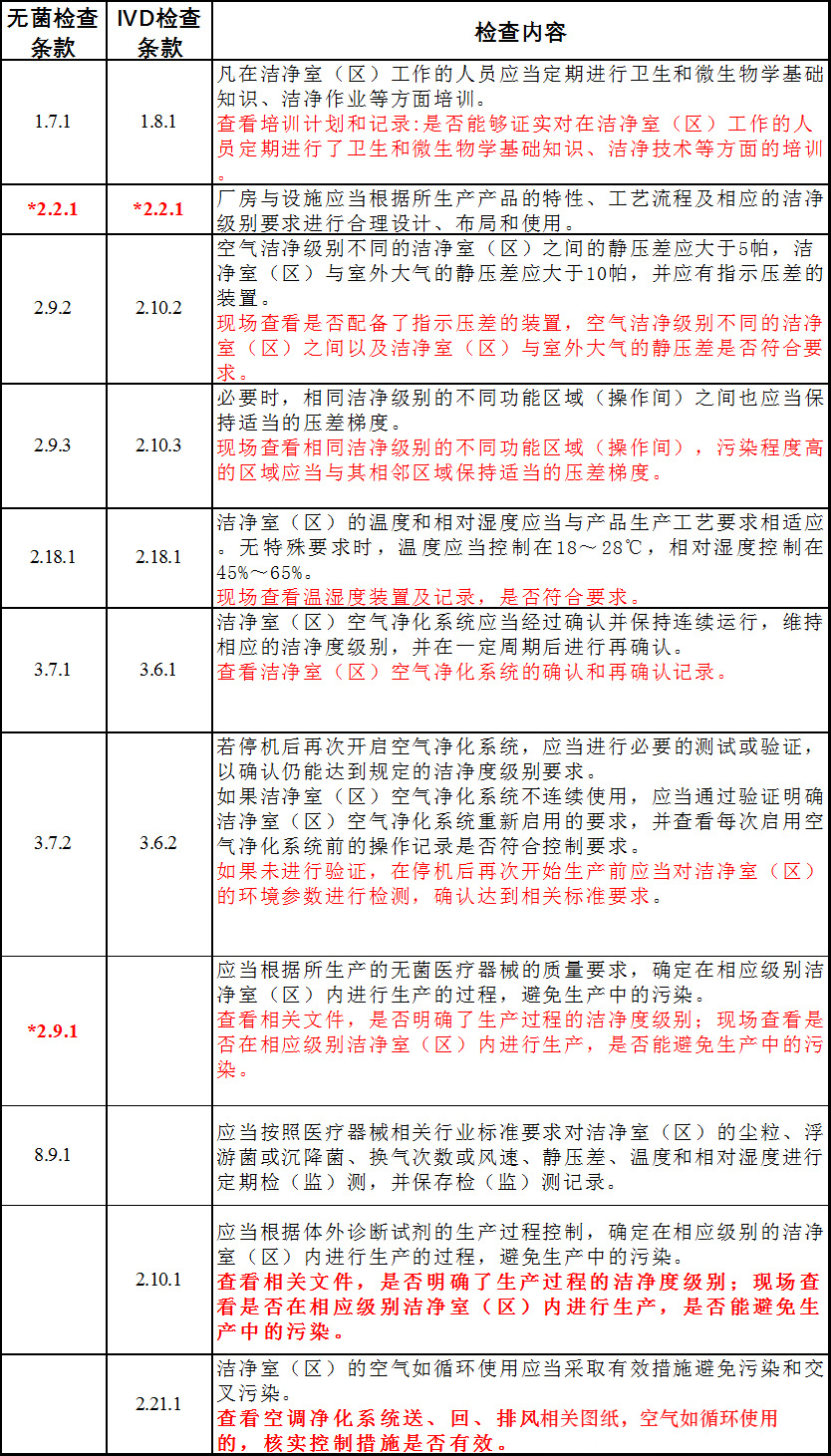
03 常见飞检不符合项
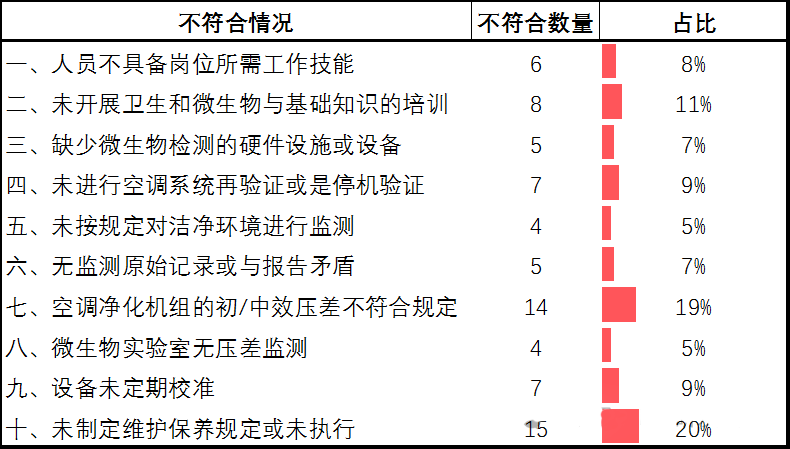
统计2016年至2022年的国家飞检不符合项,空气净化系统不符合项主要开在机构与人员、厂房设施、设备、质量控制章节。空气净化系统的不符合类型及占比如下:
医疗器械制造展Medtec展前会分论坛将隆重举办分论坛2:质量提升与体系、标准建设,本次会议将邀请行业大咖一起对行业内备受关注的质量热点内容进行解析和讨论,致力于为医疗器械法规和质量人员提供最新的标准资讯,并且制定有效的实施策略。最大限度的为从业者答疑解惑。报名参观>>>
一、人员不具备岗位所需工作技能
二、未开展卫生和微生物与基础知识的培训
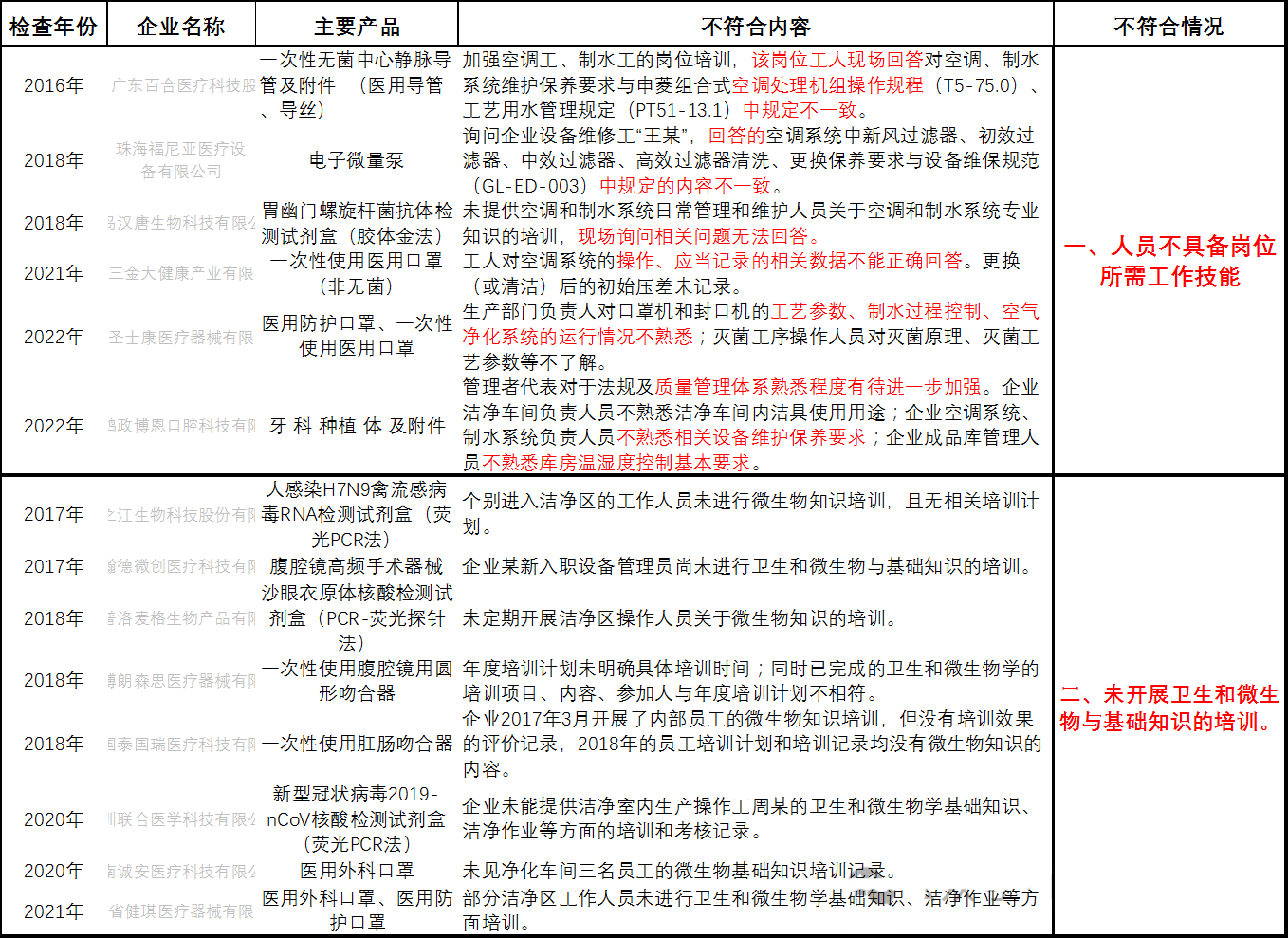
三、缺少微生物检测的硬件设施或设备
四、未进行空调系统再验证或是停机验证
五、未按规定对洁净环境进行监测
六、无监测原始记录或与报告矛盾
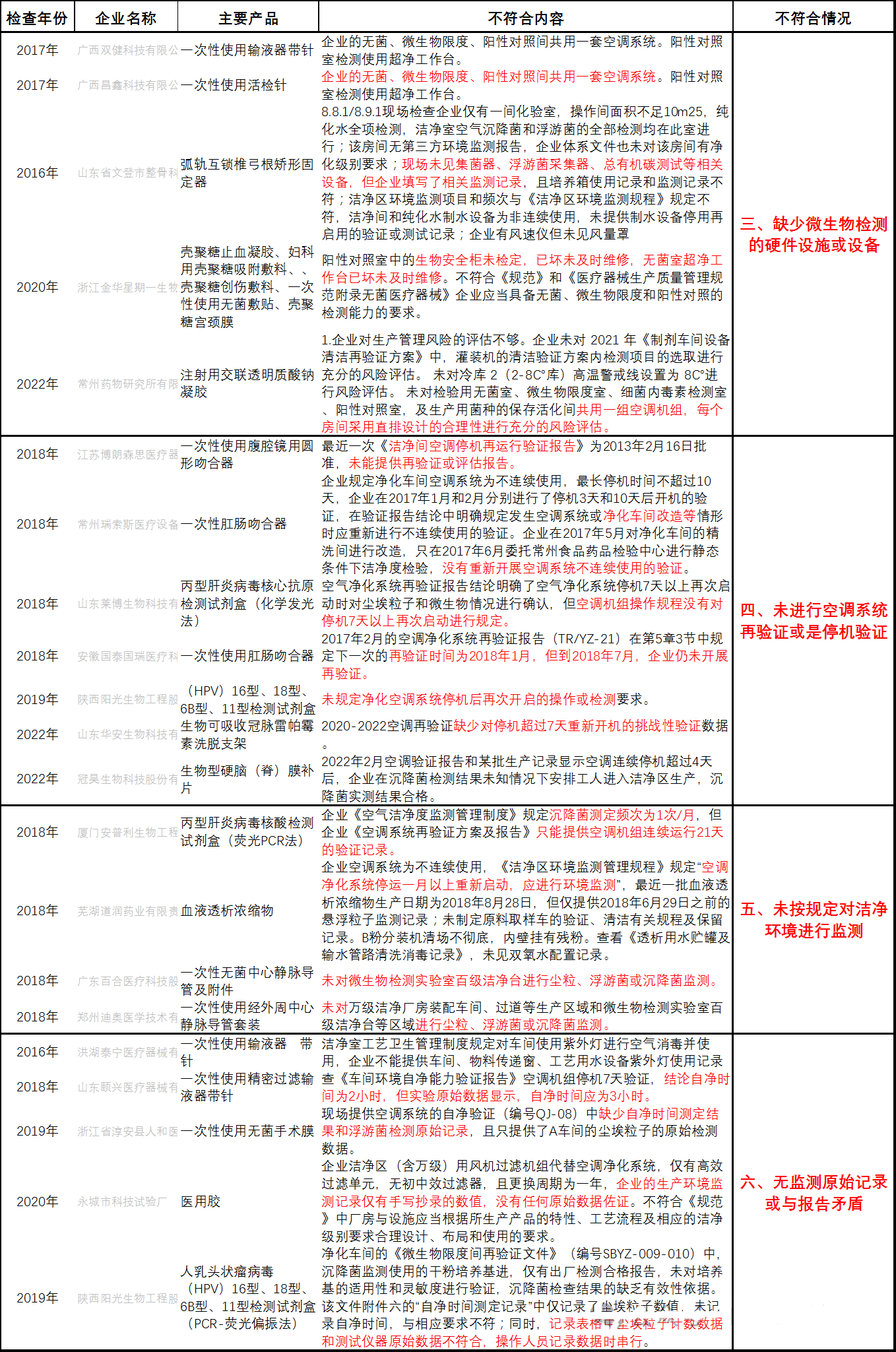
七、空调净化机组的初/中效压差不符合规定
八、微生物实验室无压差监测
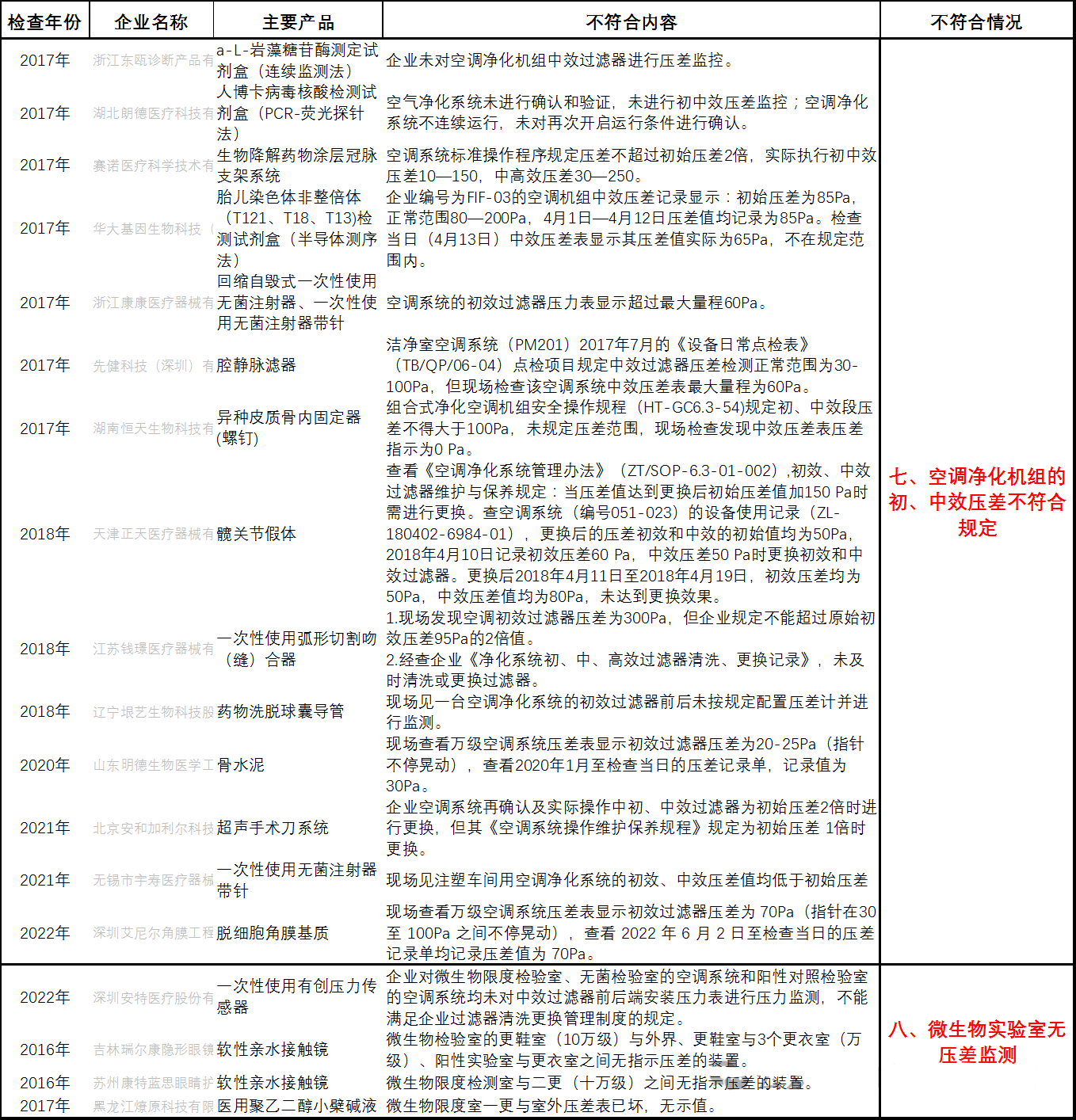
九、设备未定期校准
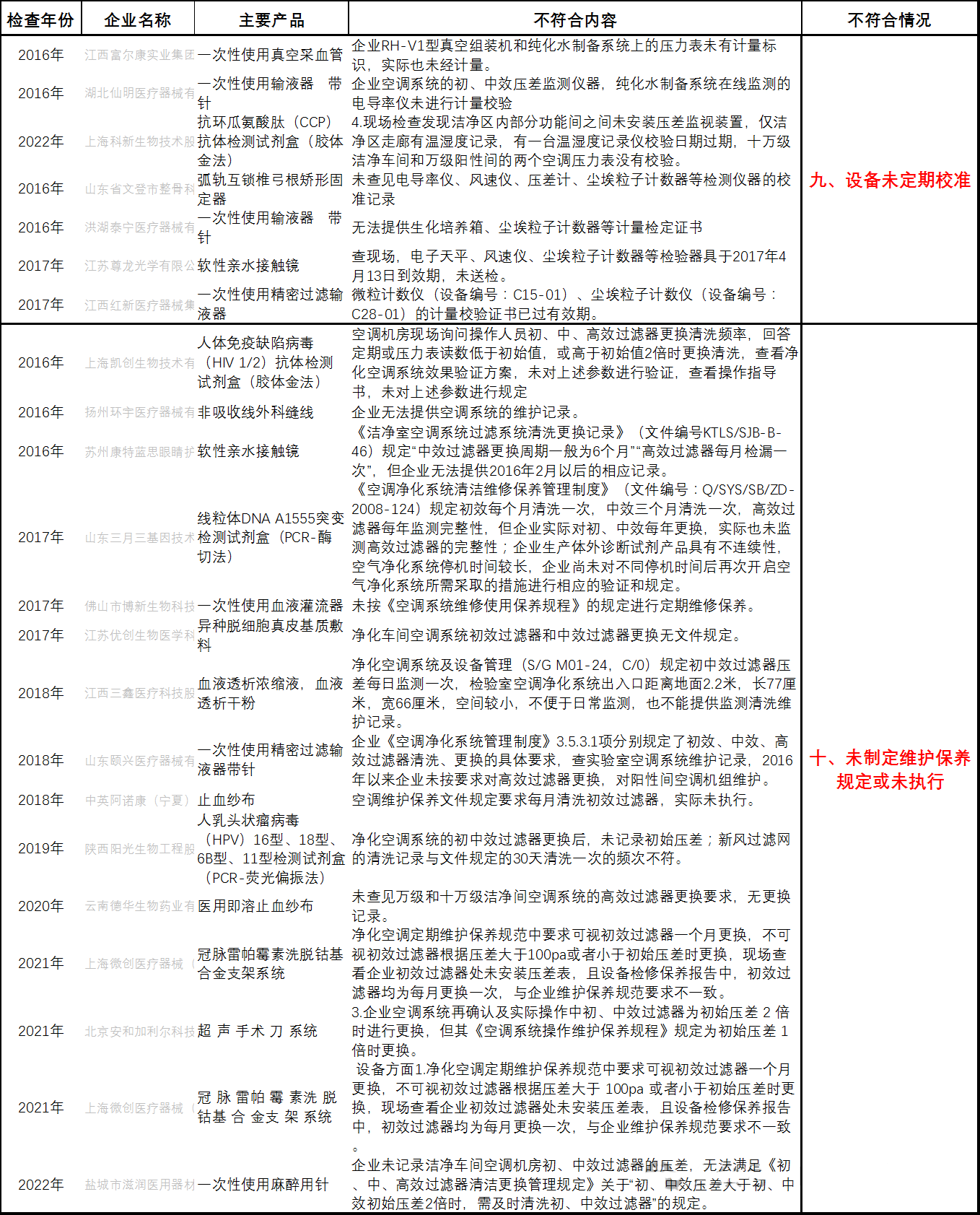
十、未制定维护保养规定或未执行
04 结语
医疗器械制造展Medtec认为对于体外诊断试剂、无菌医疗器械、植入医疗器械等产品而言,洁净区是不可缺少生产环境,洁净区空气净化系统的设计及有效运行是保证洁净度的关键之一, 也直接或间接地影响着医疗器械产品的质量。
需要具备前提条件:①需要具备专业的检验人员和维护保养人员,他们是否经过与其岗位要求相适应的培训,具有相关理论知识和实际操作技能;②是否具备环境检测设备;③阳性对照室空气净化系统是否单独设置,其室内空气是否经过过滤后直接排至室外,阳性对照室与相邻洁净区以及非洁净区压差是否符合要求。
制定合适的操作规程:①是否建立空气净化系统过滤器清洗、更换周期的制度,是否按规定对过滤器进行清洗、更换,是否进行初中效压差监控;是否监测高效过滤器的完整性;②是否建立合理可实施的环境监测操作规程。
按要求实施维保及监测:①依据维保规程实施、做好维护保养记录;②依据规程实施环境监测,监测记录完整可追溯。
按要求进行首次验证、再验证、停机验证:需要按规定进行首次验证,通过首次验证确定环境的监测频率、监测指标、维护保养频率等内容,对停机超过一定时间后进行停机验证,确定停机多久后重启需要哪些检验或验证。
文章来源:IVD 研发视野


