2024有源医疗器械创新论坛谈:ISO13485和GB/T42061解读、导入与审核(条款7.3.3 设计和开发输入)
2024-11-05
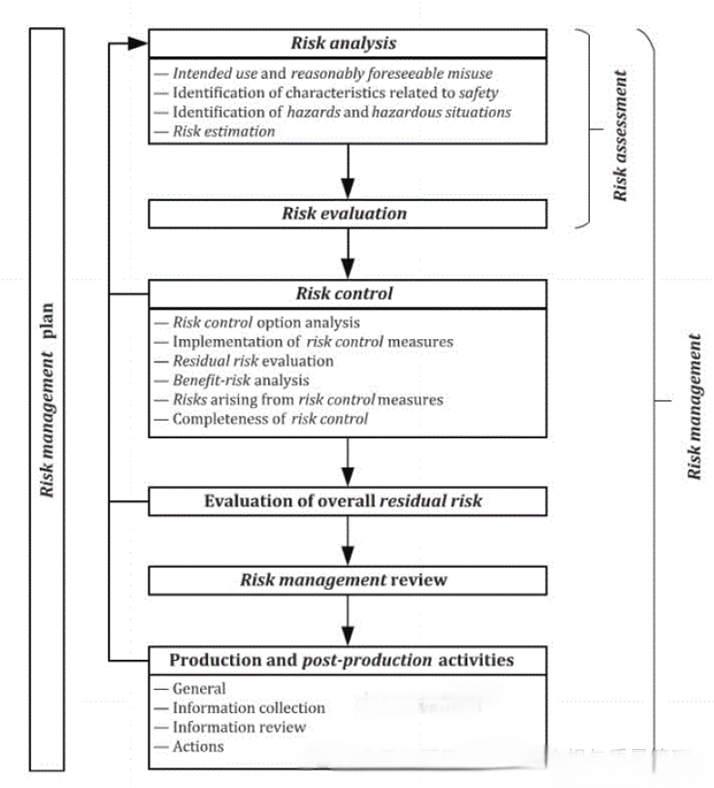
ISO13485:2016《医疗器械质量管理体系用于法规的要求》,由ISO国际标准化组织于2016年3月1 日正式发布,是一项应用于医疗器械领域的质量管理体系的国际标准,强调医疗器械的安全有效,组织提供的医疗器械要满足顾客和法规要求。
设计输入要求清晰,即设计输入应表述明确,避免模糊不清或模棱两可的要求,以确保设计团队能够准确理解并执行。
设计输入要求的可验证性,设计输入应能够被验证,即通过某种方式确认其正确性和可行性。这可能包括通过计算、模拟、测试等方法来验证设计输入是否符合要求。设计输入要求不能相互矛盾,即一致性,设计输入之间应相互协调。如果发现矛盾,应及时解决,以确保设计过程的顺利进行。
这些是确保设计质量和医疗器械产品成功的关键因素。
-疾病的诊断、预防、监护、治疗或缓解;
-损伤的诊断、监护、治疗、缓解或补偿;
-生理结构或生理过程的查验、替代、调节或支持;
-生命的支持或维持;
-妊娠控制;
-医疗器械的消毒;
-通过对取自人体的样本进行体外检查的方式来提供信息。其作用于人体体内或体表,主要预期功能不是通过药理学、免疫学或代谢的方式实现,但这些方式有助于实现预期功能。