2025医疗器械展会Medtec一文读懂美国FDA和欧盟MDR医疗器械法规异同!
2025-01-02
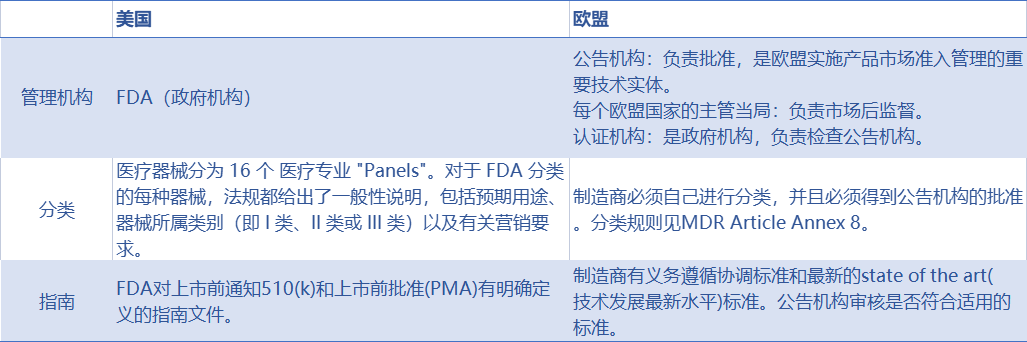
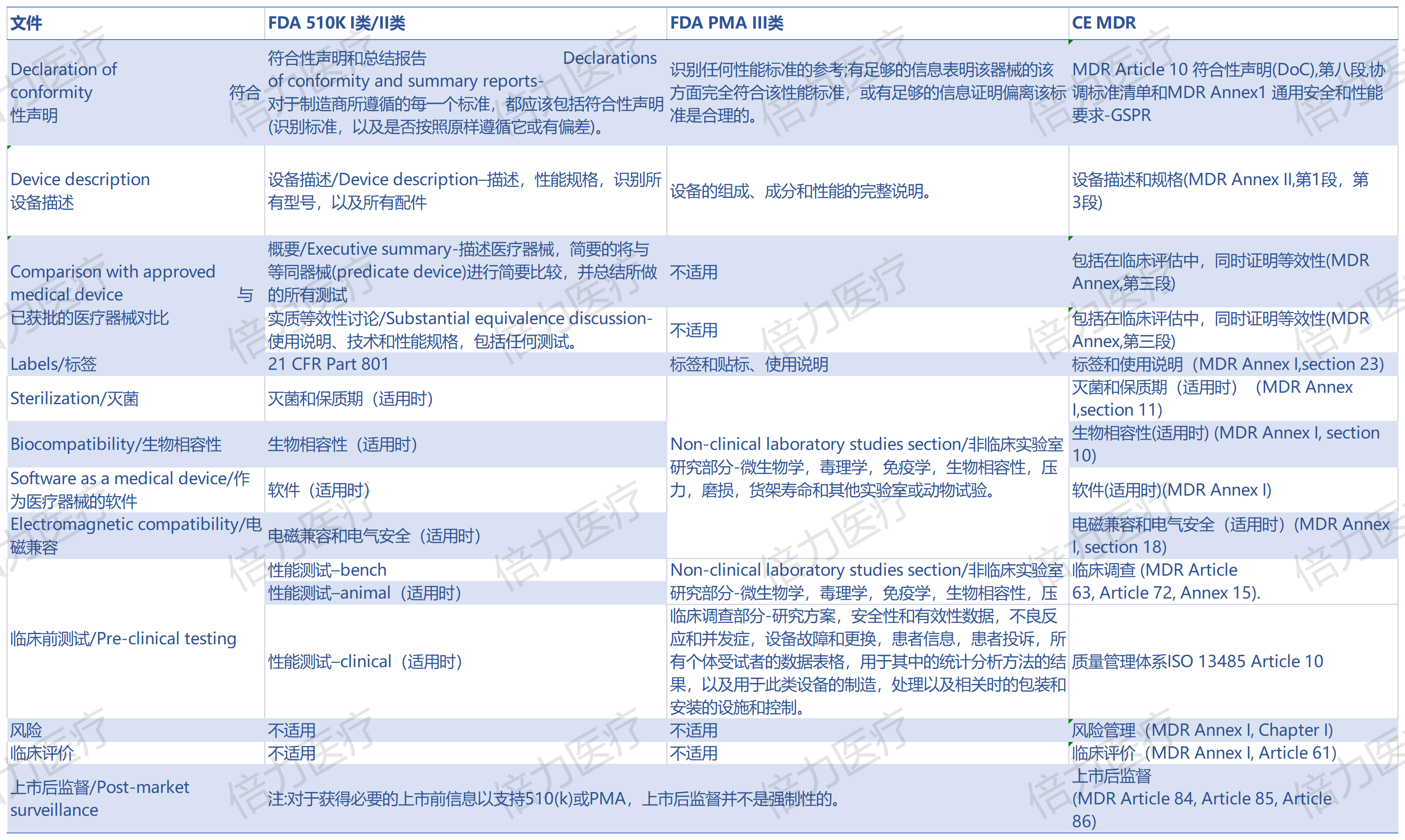
2025医疗器械展会Medtec指出,而器械制造商对于目标市场地区的监管法令是一定要做足功课的。但与此同时,地区与地区之间在监管法令方面势必会存在一些差异,这也就需要制造商们“对号入座”,选择适合自己的途径,以及合理避开一些障碍。近期,随着最近欧盟采用MDR之后,FDA和欧盟法规现在比以前更加匹配。在质量体系要求(均符合 ISO 13485)、合格评定先决条件和符合协调标准(SaMD 的IMDRF 标准就是其中之一)方面尤其如此。

尽管是像“兄弟”,二者仍存在一些差别:

2025医疗器械展会Medtec现场将设四大专区,囊括电子光学、AI、IVD诊断及影像、检测、临床、法规、研发设计& 大动物实验专区等展品,点击此处报名参展>>>