







国际医疗器械设计与制造技术展览会独家报导植入式医疗器械和相关的欧盟MDR要求
2023-07-27
植入式医疗器械是一种埋置在生物体或人体内的电子设备,具有重要的医疗作用;植入式医疗器械能够将患者数据传送给WBAN(无线个人网)中的体外设备然后通过移动互联网传送到大型医院、专业医疗中心等平台上,为糖尿病、高血压、心脏病等各种慢性疾病患者提供远程的监测与诊断,也为科学的治疗方案的制定提供了数据支撑。
植入式医疗器械通常是高风险医疗器械,其潜在故障可能会给患者带来严重后果。这就是监管机构高度关注此类医疗器械安全性的原因——特别是在新欧盟医疗器械法规2017/745中。
在本文中,我们将根据欧盟法规介绍与植入式医疗器械相关的主要要求。
鉴于这些类型设备的特殊性,有一些特定要求需要考虑,并应在医疗器械文件或其他类型的文件中提供合规性证据,例如在法规遵从策略背景下。
什么是植入式医疗器械?
欧盟MDR文本中规定的植入式医疗器械定义如下:
“植入式器械”是指部分或完全吸附的医疗器械,其目的是:
-
完全进入人体或
-
借助临床手术替代人体上皮表面或眼表面,并且在手术后保留在人体内。
任何借助临床手术部分进入人体内,并且在手术后留在人体内至少30天的医疗器械也应被视为植入式器械。
植入式器械的一般考虑因素
与其他医疗器械一样,植入式医疗器械可以分为有源器械和无源器械。无源器械内部没有能量源,并且可以通过其他方式激活,如患者的运动或呼吸。
而有源植入式医疗器械是通过自然腔道(口)或手术方式插入患者体内的通电设备,并在手术后保留在患者体内。
有源植入式医疗器械被认为是高风险器械,需要实现高标准制造和更高水平的法规合规性。
ISO 13485要求
ISO 13485:2016包含与植入式医疗器械相关的多项要求,汇总如下图所示。我们将首先了解主要要求,然后再转向欧盟MDR 2017/745中的规定要求。
植入式医疗器械
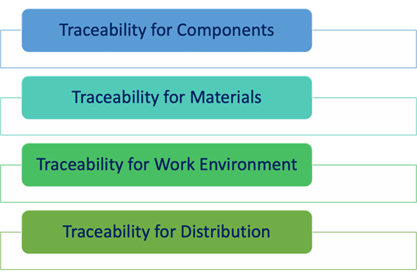
第7.5.9.2节规定了可追溯性方面的特定要求。具体而言,应保留以下方面的可追溯性记录:
-
部件。重要的是,如果医疗器械由不同的部件成,则应保持所有这些部件的可追溯性。基本上,每个部件都应与特定BOM和特定的生产记录关联。如此,在任何时候都可以追溯到指定医疗器械中包含的部件。
-
材料。用于制造植入式医疗器械的所有材料都应被正确识别和追踪;应提供材料适合用于此类医疗器械的具体证明,因为这对于患者的整体安全至关重要。
-
工作环境。显而易见的是工作环境对于植入式医疗器械的影响是巨大的,特定工作条件可能会直接影响器械的质量和安全。因此,有必要准确记录工作环境条件,如温度、湿度等。
此外,植入式医疗器械的可追溯性附加要求如下:
机构/企业应要求分销服务供应商或分销商保存医疗器械的分销记录,以实现可追溯性,并确保这些记录可供检查。
这与分销服务供应商或分销商有关,他们有义务保存医疗器械的分销记录。这点至关重要,因为始终有必要了解最终患者,以便在发现任何安全问题时可以与患者快速联系。
最后,ISO 13485:2016第8.2.6节中还提出了对植入式医疗器械的另一项要求,其中明确规定必须记录在产品发布过程中进行医疗器械检查或测试的所有人员的身份。
Medtec China已经从2012年开始连续举办了8届植入介入医疗器械峰会,会议围绕骨科植入物、心血管介入产品,探讨其法规政策、市场趋势、研发与设计与材料创新等内容,为植入介入制造商材料供应商及服务商等提供一个讲解公司新技术及材料的平台。扫描下方二维码立刻报名参展加入我们~

欧盟MDR 107/745要求
根据EU MDR 2017/745,与植入式医疗器械相关的最重要的新要求是所谓的植入卡。该要求旨在改善与患者共享的植入式医疗器械的相关信息。
植入卡有不同的用途,例如:
-
使患者能够识别植入的医疗器械并获取安全相关信息。
-
使患者能够在特定情况下识别自己是需要特殊护理的人员,例如安全检查或其他情况。
该法规第18条定义了植入卡的相关要求,其中明确规定了植入卡中需要包含的信息。具体而言,应包括以下信息:
-
器械名称
-
器械类型
-
唯一器械标识(UDI)– DI和PI均应包含在内
-
序列号或批号(如适用)
-
医疗器械制造商的名称和地址
-
医疗器械制造商的网站
-
患者或医疗专业人员针对可合理预见的外部影响、医疗检查或环境条件的相互干扰,所应采取的任何警告、预防措施或纠正措施,如第18(b)条所述。
-
医疗器械的预期使用寿命
-
确保患者安全使用器械的其他信息
此外,还需要包含患者的信息。包括患者姓名、植入日期和进行器械植入的医疗机构。这些信息应在植入卡交给患者时就包含在内,以便尽可能地尊重隐私和GDPR要求。
此外,植入卡还有其他具体要求,如下图中所述:

我们将不会详细介绍所有这些具体要求,但很重要的一点是,MDCG(欧盟医疗器械协调小组)发布了一份具体指南MDCG 2019-08,其中说明了与植入卡相关的所有要求。
植入式医疗器械相关的ISO标准
在产品设计、生产和后期制造阶段,还需要适当考虑与植入式医疗器械相关的其他标准。下面是我们找到的与植入式医疗器械相关的最重要的ISO标准清单;但这并不是详尽清单,根据设备的类型,其他ISO标准也可能适用。
-
ISO 14708-1:2014 – 安全、标记和制造商所提供信息的通用要求
-
ISO 14708-2:2012 – 心脏起搏器
-
ISO 14708-3:2017 – 植入式神经刺激器
-
ISO 14708-4:2008 – 植入式输液泵
-
ISO 14708-5:2010 – 循环支持器械
-
ISO 14708-6:2010 – 治疗快速心律失常的有源植入式医疗器械的特殊要求(包括植入式心脏除颤器)
-
ISO 14708-7:2013 – 人工耳蜗系统的特殊要求
-
ISO 14117:2012 – 植入式心脏起搏器、植入式心脏复律除颤器和心脏再同步器械的电磁兼容性测试协议
-
ISO 14602:2010 – 无源外科植入物 – 骨接合植入物 – 特殊要求
-
ISO 14630:2012 – 无源外科植入物 – 通用要求
-
EC 62304:2006 – 医疗器械软件
技术文件的合规性检查清单
为了便于在向公告机构提交档案之前控制技术文件的合规性,QualityMedDev根据MDR 2017/745编制了一份合规性检查清单,其中详细列出了对技术文件的所有要求。
该技术文件检查清单将作为简化技术档案符合性评估的重要工具。这份15页的检查清单以Word文件形式提供,因此是完全可编辑的,可适应贵组织推向市场的产品类型。

文章来源:Medtec医疗器械设计与制造


