在药品生产过程中,发生变化是非常正常的。即使在R&D能力相对较高的发达国家,每年控制一个品种在5-10个之间的变化也是很常见的,如果涉及到新产品、无菌产品、生物制品等。数量会更多。而在国内,目前对变化的控制还达不到国外的先进水平。如果控制执行不力,药品质量安全的风险系数会大大增加。近年来,国内医药行业爆发的“药害”事件,很多都是因为生产中的变更控制不力!
1.前言
根据设计变更的影响程度,可分为重大变更、中等变更、微小变更;根据是否需要向监管机构注册、备案、报告,又分为变更注册、变更备案、变更报告。在2021年6月1日实施新的《医疗器械监督管理条例》之前,变更注册是指许可事项变更,而变更备案是指登记事项变更。
设计变更分类也是依据对产品安全性、有效性影响的程度进行划分和管理,基于变更可能引入的风险进行评估,类似医疗器械产品根据产品风险程度分为一类、二类、三类进行监管。
医疗器械产品的分类比较清晰,一是可以依据医疗器械分类规则判断,二是可以根据医疗器械分类目录查找,三是有明确的分类界定流程可走。更多医疗器械法规以及监管条例解读,请关注上海医疗器械展会Medtec China,会议议题覆盖新修订的《医疗器械监督管理条例》及相关配套新政览读、医疗器械质量管理信息化、有源医疗器械临床评价常见问题、医疗器械突发事件应急处置与媒体沟通演练。点击快速加入上海医疗器械展会Medtec China。
设计变更分类的指导性文件较少,第一原则是依据《医疗器械注册与备案管理办法》条款进行判断;然后再根据具体的规范性文件进行确认和开展,如《医疗器械说明书和标签管理规定》、《总局办公厅关于体外诊断试剂说明书文字性变更有关问题的通知》、《医疗器械注册单元划分指导原则》、《医疗器械软件注册技术审查指导原则》、《体外诊断试剂变更注册审查指导原则》等。
2.变更控制咨询
通过质量管理体系程序控制设计变更时,工作的流程、涉及的人员、工作的内容与产品首次设计开发有绝大多数的重叠;而对外走好合规路径审批流程,需要理清相关法规文件,根据法规文件无法确认时,需要同监管机构沟通。
上海医疗器械展会Medtec China提醒,由于注册人和监管机构双方是陌生的,角色差异大,沟通难度自然比部门内部、跨部门间要大,沟通需要做好充分准备。
首先要找对的人:了解国家局、省局的组织架构和各部门职能,才能找到对的人,监管部门信息现在也非常透明,绝大多数信息会主动公开,直接在官网就可以找到。
其次要了解问题:对问题的背景进行充分调查、分析、内部形成基本的方案,逐步把问题聚焦到非常窄的范围,问题太开放,在有限的沟通时间内很难得到答案。
最后要换位思考:了解监管的目的和角色,原则性问题无需要讨论,要比监管方更加清楚法规要求和风险隐患,不要把监管方当咨询师,而是汇报对象,多说少问,多领导少附和。
3.共性问题回复
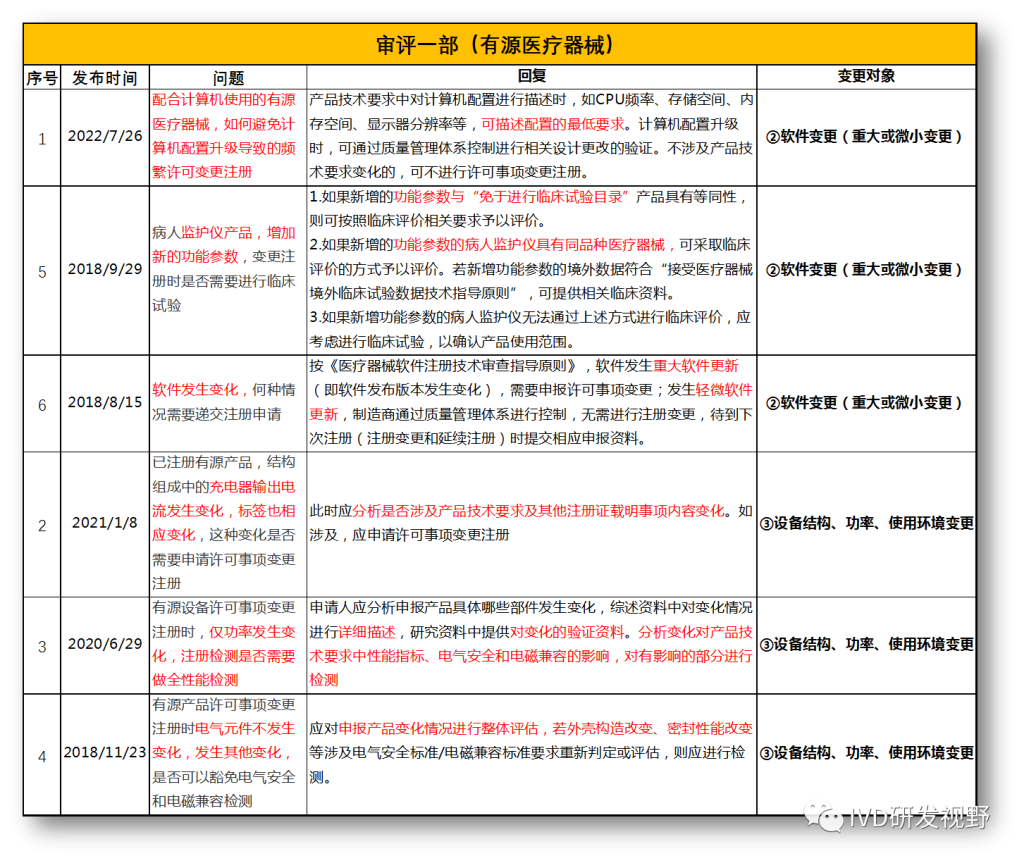
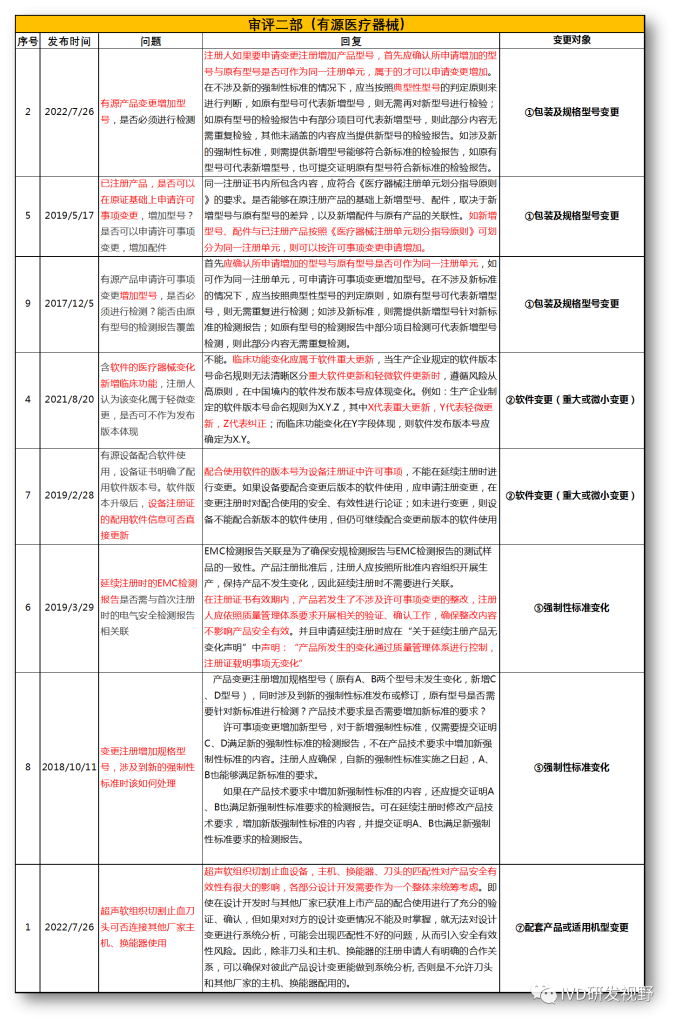
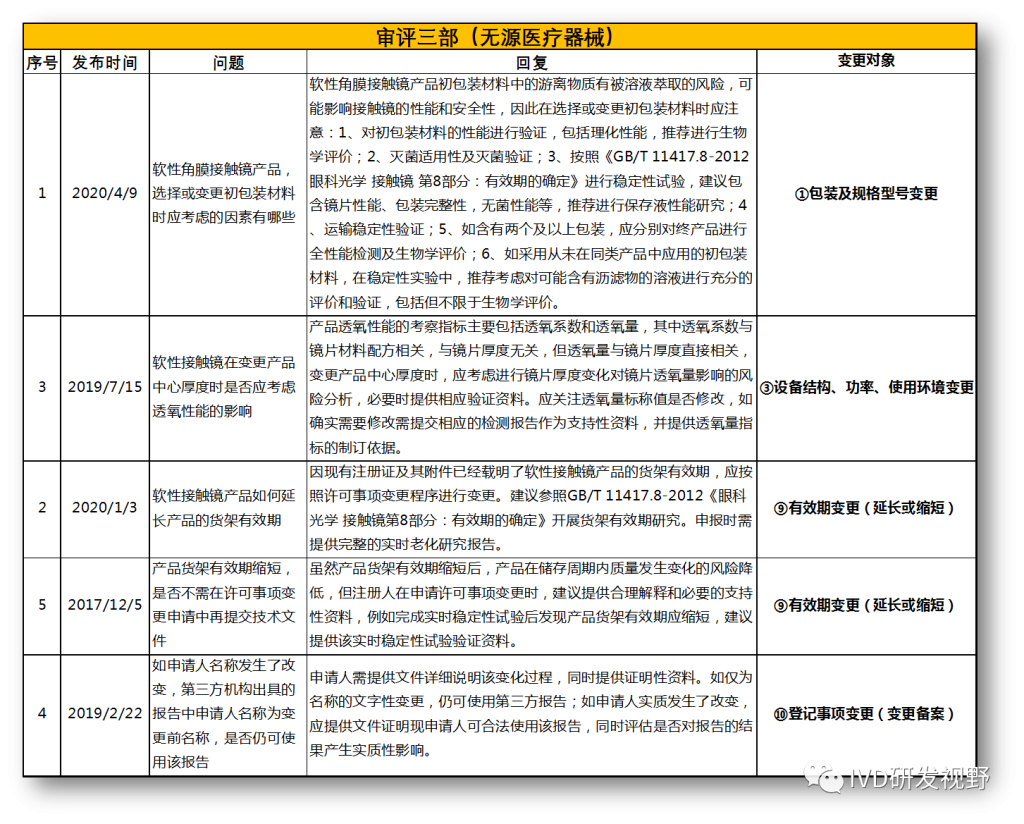
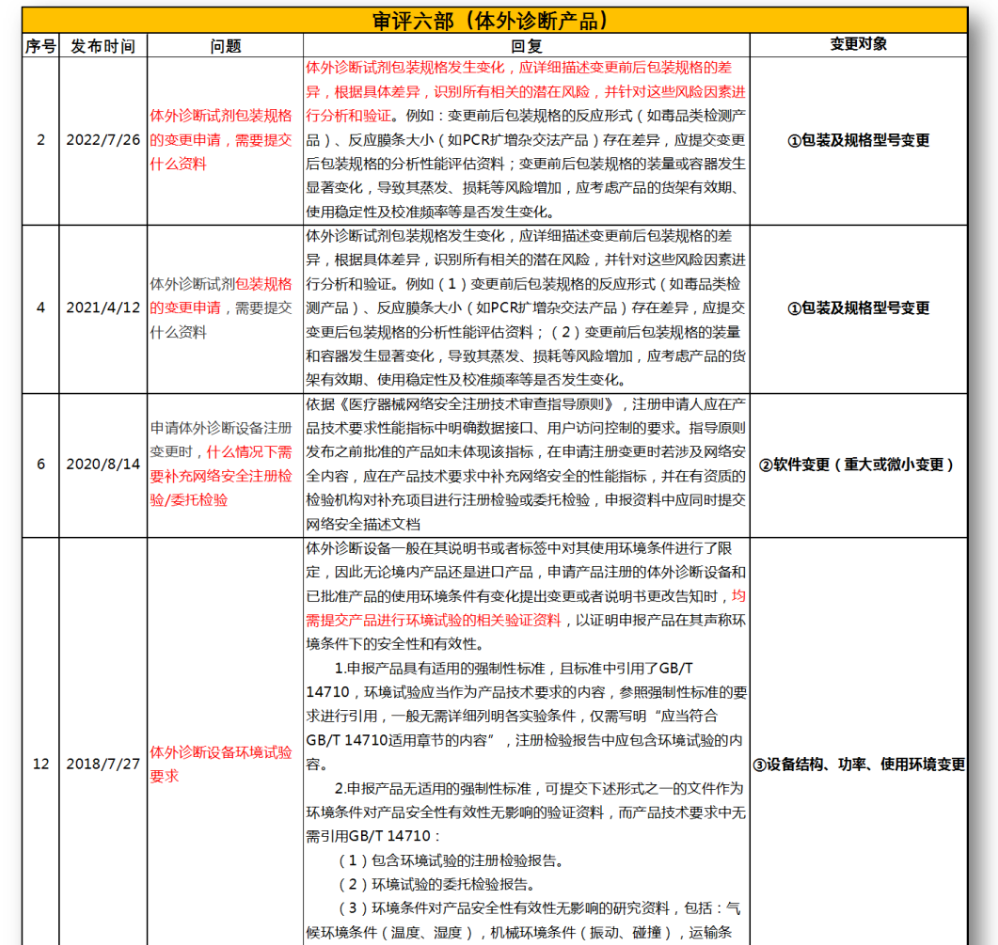
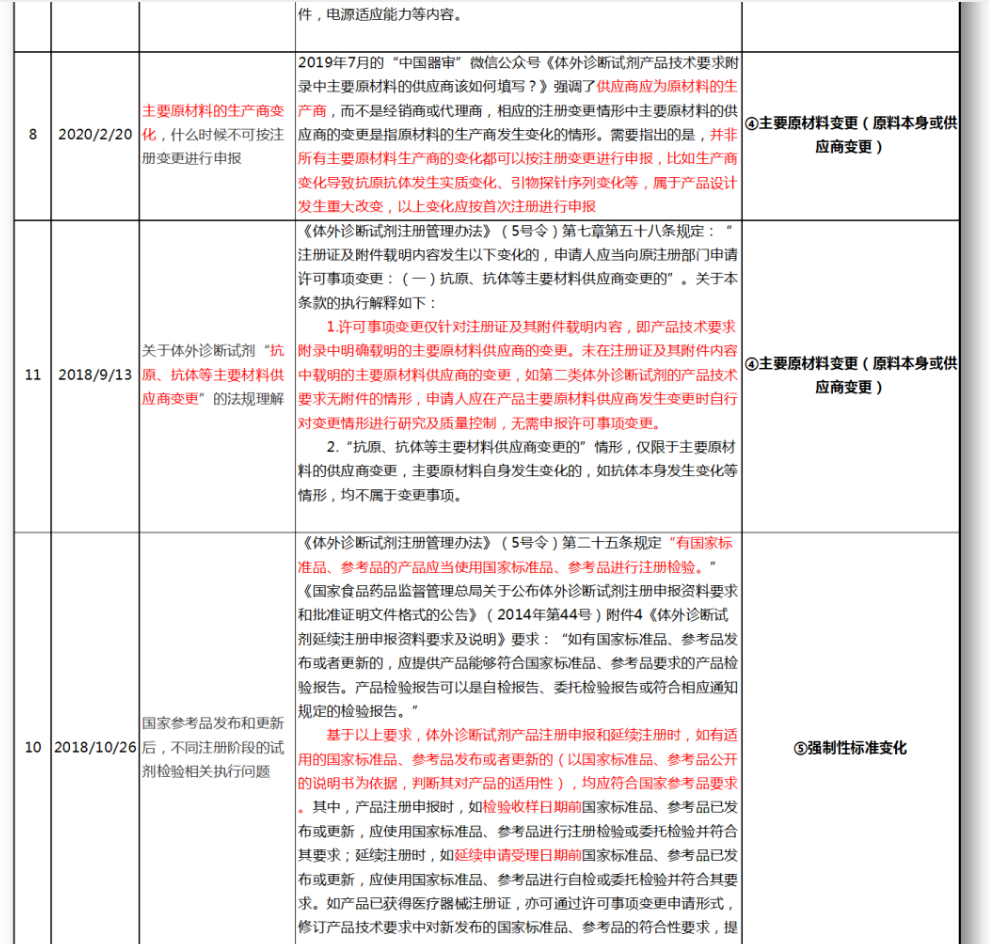
无法明确的变更种类,在沟通前先了解是否有同类问题,并且审评中心已给出回复,以下是国家医疗器械技术审评中心关于变更的共性问题回复,主要集中在以下几种类型。
①包装及规格型号变更
②软件变更(重大或微小变更)
③设备结构、功率、使用环境变更
④主要原材料变更(原料本身或供应商变更)
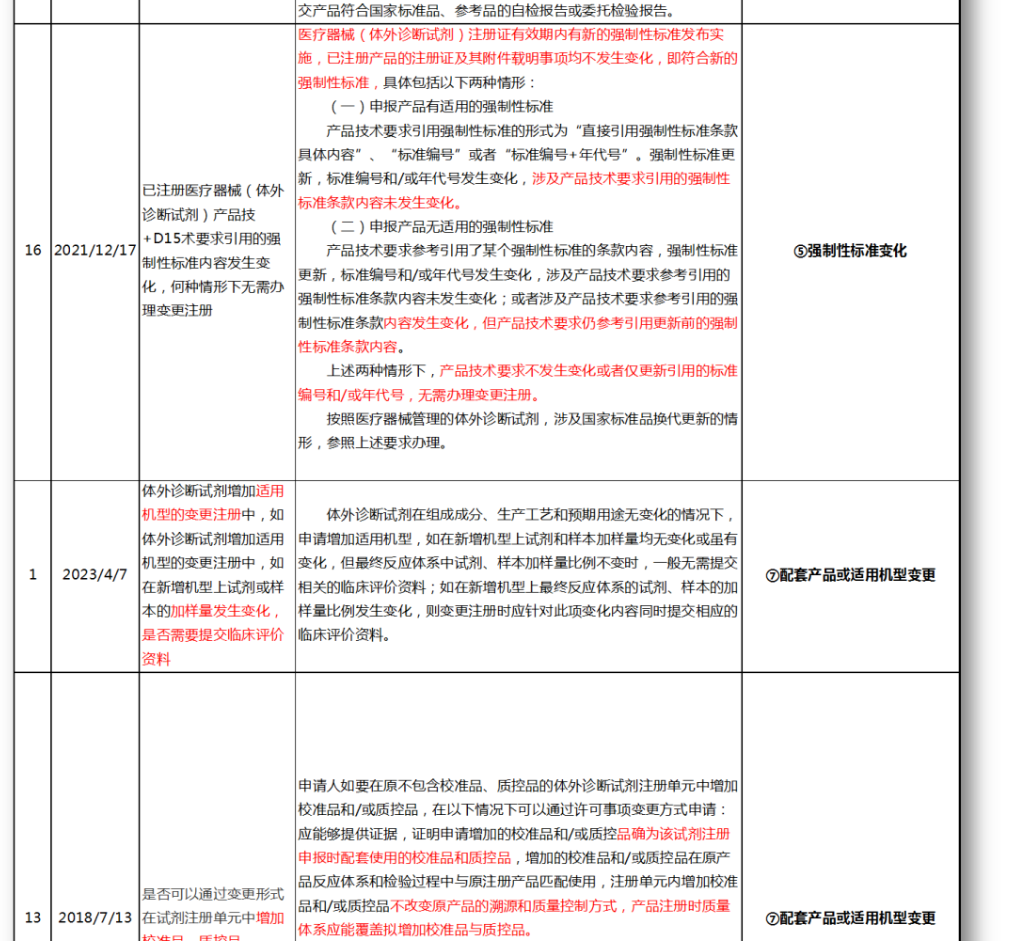
⑤强制性标准变化
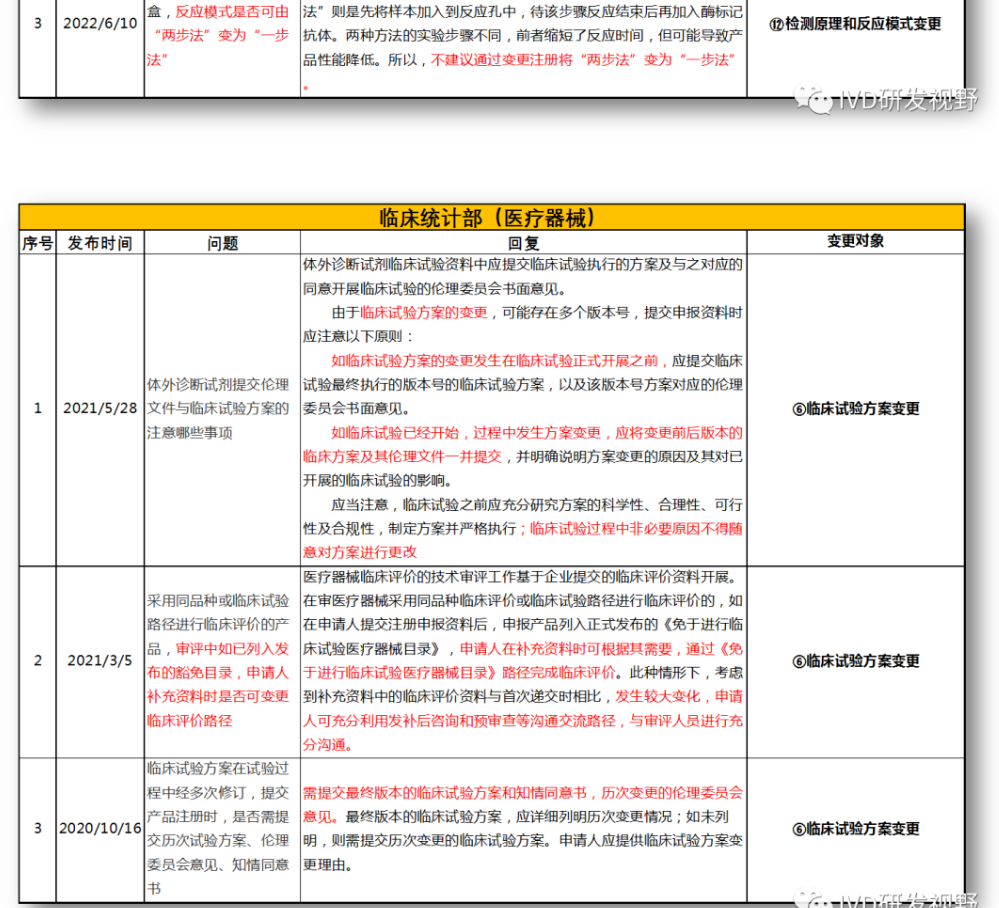
⑥临床试验方案变更
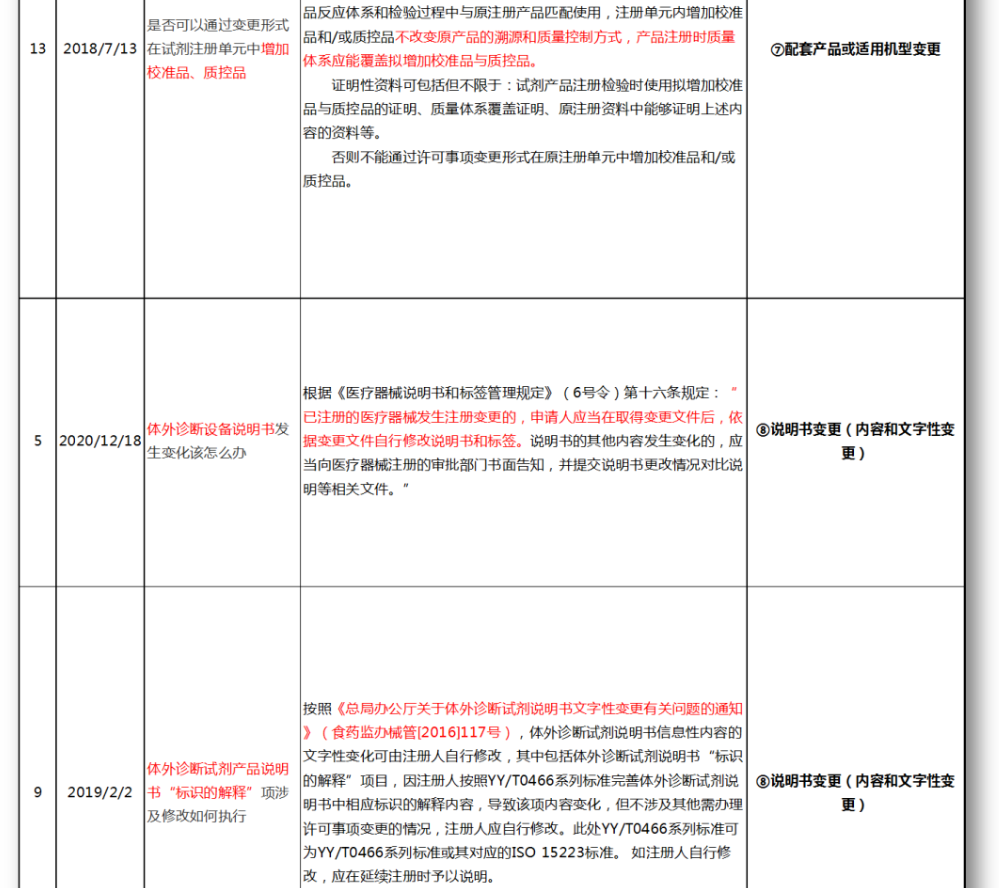
⑦配套产品或适用机型变更
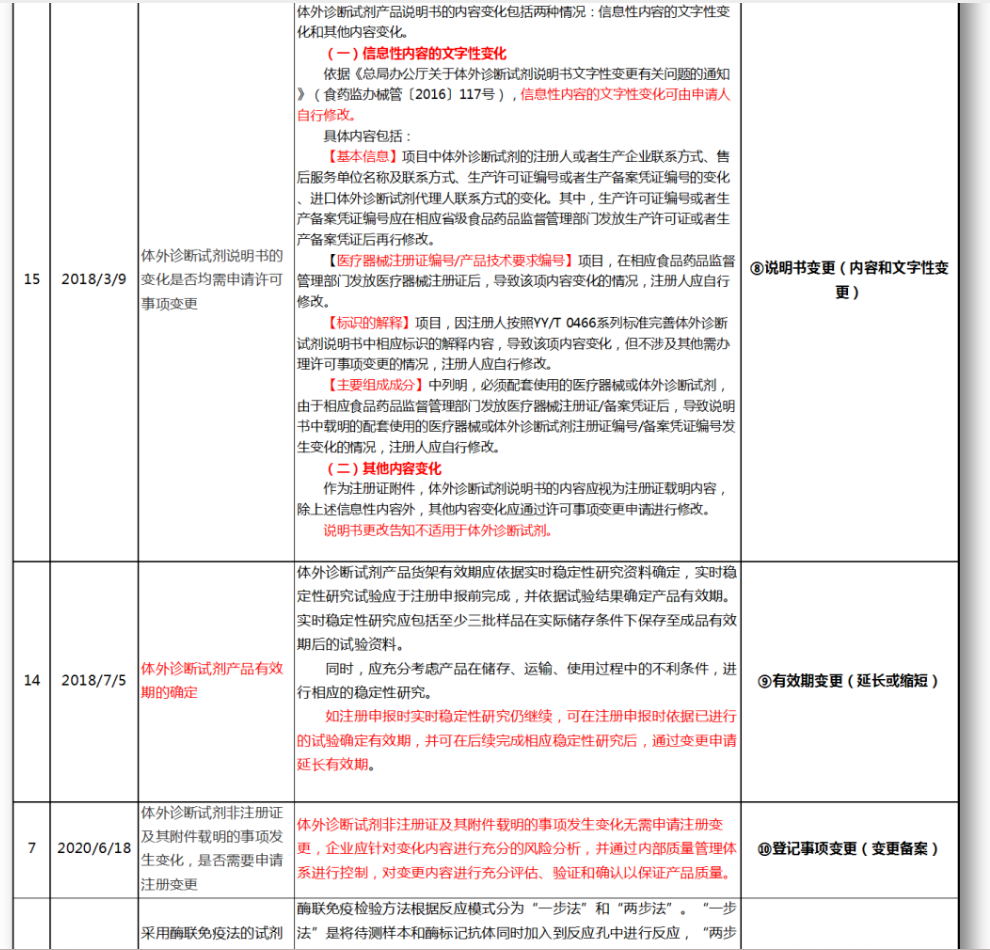
⑧说明书变更(内容和文字性变更)
⑨有效期变更(延长或缩短)
⑩登记事项变更(变更备案)
⑪灭菌工艺变更
⑫检测原理和反应模式变更